02 Working with larger VCF: T2T by chromosome
Vincent J. Carey, stvjc at channing.harvard.edu
October 30, 2024
Source:vignettes/large_t2t.Rmd
large_t2t.RmdOverview
In this document we illustrate some issues with large data volumes.
Here is how we acquire the genotypes for the thousand genomes samples based on the T2T reference. We obtained bgzipped vcf via
AnVIL::gsutil_cp("gs://fc-47de7dae-e8e6-429c-b760-b4ba49136eee/1KGP/joint_genotyping/joint_vcfs/raw/chr22.genotyped.vcf.gz", ".")Then we used hail in python to deal with the conversion to MatrixTable. We could have done this in R, but we had to learn how to manipulate the ‘reference sequence’. We have a conjectural ‘reference sequence’ json document in the json folder of the BiocHail package, used here as t2tAnVIL.json.
>>> import hail as h
>>> rg = h.get_reference('GRCh38')
Initializing Hail with default parameters...
Setting default log level to "WARN".
To adjust logging level use sc.setLogLevel(newLevel). For SparkR, use setLogLevel(newLevel).
Running on Apache Spark version 3.1.3
SparkUI available at http://756809c79837:4040
Welcome to
__ __ <>__
/ /_/ /__ __/ /
/ __ / _ `/ / /
/_/ /_/\_,_/_/_/ version 0.2.105-acd89e80c345
LOGGING: writing to /home/rstudio/hail-20221213-1558-0.2.105-acd89e80c345.log
>>> nn = rg.read("t2tAnVIL.json")
>>> h.import_vcf("chr22.genotyped.vcf.gz", force_bgz=True, reference_genome=nn).write("t2t22.mt")This operation seems to take a long time even with 64 cores. We could not get exact timing owing to some connectivity problems.
Here we’ll work with the genotype data for T2T chr17. We assume that the MatrixTable is located at the path given by the environment variable HAIL_T2T_CHR17. This MatrixTable is available in the Open Storage Network at osn:/bir190004-bucket01/Bioc1KGt2t/t17.zip. This is a 45 GB file. It should be unzipped at the location pointed to by HAIL_T2T_CHR17. Instructions for using rclone to acquire the zip file are given in the appendix.
(If the above command indicates that BiocHail is not available, see the Installation section near the end of this document.)
hl <- hail_init()
# NB the following two commands are now encapsulated in the rg_update function
nn <- hl$get_reference('GRCh38')
nn <- nn$read(system.file("json/t2tAnVIL.json", package="BiocHail"))
# updates the hail reference genome
bigloc = Sys.getenv("HAIL_T2T_CHR17")
if (nchar(bigloc)>0) {
mt17 <- hl$read_matrix_table(Sys.getenv("HAIL_T2T_CHR17"))
mt17$count()
}Population stratification assessment via PCA
The following command would compute PCs based on all SNP.
pcastuff = hl$hwe_normalized_pca(mt17$GT)This seems impractical. We have found that with a 64 core machine at terra.bio, PCA on samples of 1-5% of the 3.8 million loci on T2T chr17 takes 40 minutes. Hail’s code seems good at utilizing all the cores.
We saved the PC scores for PCA based on 38k randomly sampled loci, and 191k randomly sampled loci. Here’s a simple view of the latter.
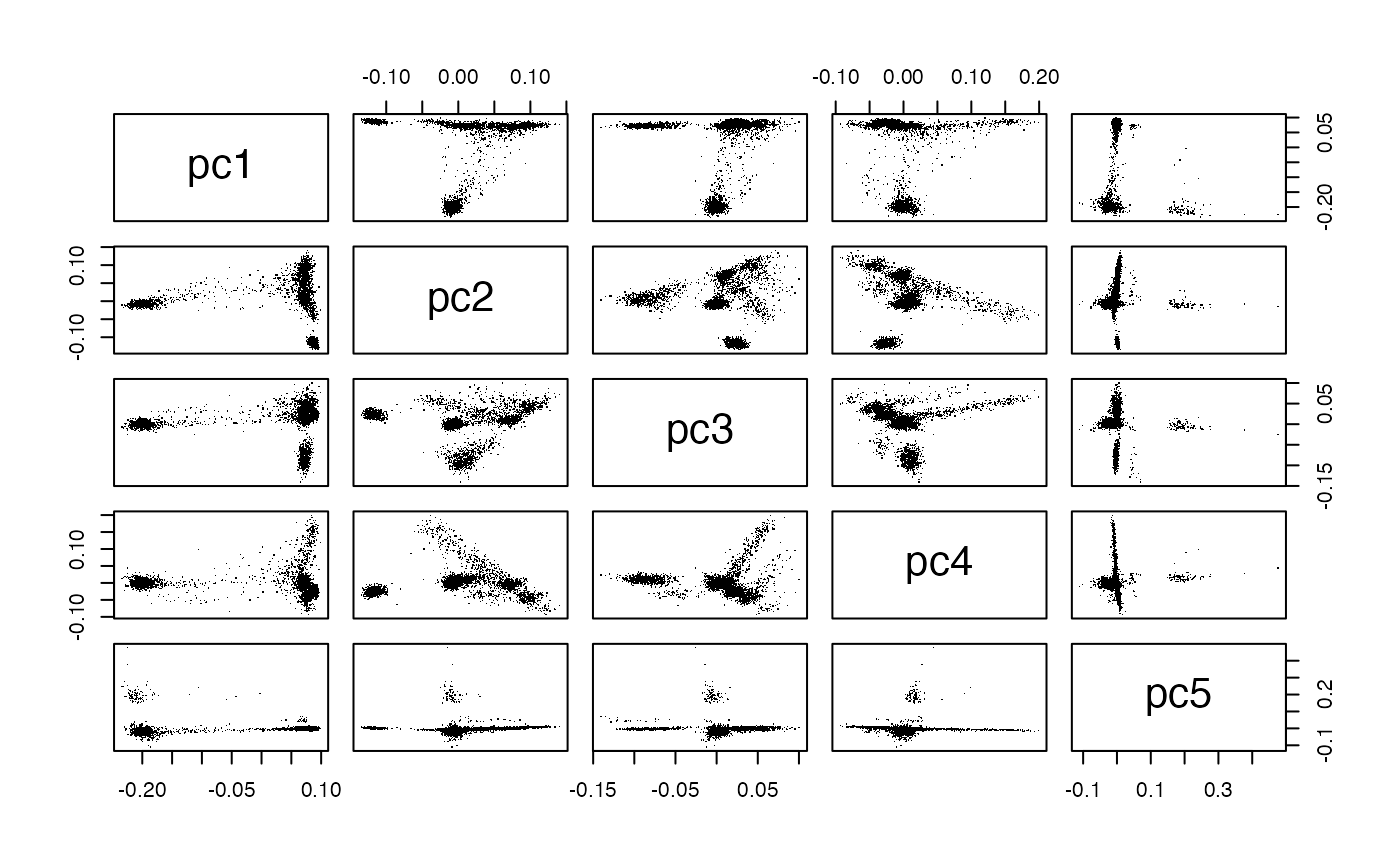
Exercises
Comment on the gain in information about geographic origin achieved by using a 5% sample of loci instead of a 1% sample.
Find the geographic origins of donors of the 1000 genomes genotypes and bind them to
mt17using the methods given in vignette01_gwas_tut. Use these codes to color the points in the PCA plot.Produce an artificial “phenotype” for these donors via
rnorm(3202,0,1), bind it to the genotype data, and perform a naive GWAS. What are the loci most strongly associated with the artificial phenotype?Produce a new artificial phenotype which has some association with geographic origin of donor, but no association with genotype. Produce a new naive GWAS, and a third using some of the PCA scores as covariates. What are the effects of this covariate adjustment on reasoning about genetic association with the artificial phenotype?
Appendix: using rclone with docker to get the chr17 data
It can be painful to install and configure rclone. We use a docker container. Let RC_DATADIR be an environment variable evaluating to an available folder.
Also, place the text file with contents
[osn]
type = s3
provider = AWS
endpoint = https://mghp.osn.xsede.org
acl = public
no_check_bucket = truein a file rclone.conf in a folder pointed to by the environment variable RC_CONFDIR.
Then the following
docker run -v $RC_DATADIR:/data -v $RC_CONFDIR:/config/rclone -t rclone/rclone:latest ls osn:/bir190004-bucket01/Bioc1KGt2twill list the files with 1KG samples genotyped against the T2T reference.
Use the rclone copyto command to obtain a local copy of the zip file t17.zip in the folder pointed to by $RC_DATADIR:
docker run -v $RC_DATADIR:/data -v $RC_CONFDIR:/config/rclone -t rclone/rclone:latest copyto osn:/bir190004-bucket01/Bioc1KGt2t/t17.zip ./t17.zipThis file should be unzipped in a folder to which the environment variable HAIL_T2T_CHR17 will point.
Installing BiocHail
BiocHail should be installed as follows:
if (!require("BiocManager"))
install.packages("BiocManager")
BiocManager::install("BiocHail")As of 1.0.0, a JDK for Java version <= 11.0 is necessary to use the version of Hail that is installed with the package. This package should be usable on MacOS with suitable java support. If Java version >= 8.x is used, warnings from Apache Spark may be observed. To the best of our knowledge the conditions to which the warnings pertain do not affect program performance.
SessionInfo
## R version 4.4.1 (2024-06-14)
## Platform: aarch64-apple-darwin20
## Running under: macOS Sonoma 14.7
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/New_York
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BiocHail_1.7.0 BiocStyle_2.32.1
##
## loaded via a namespace (and not attached):
## [1] sass_0.4.9 utf8_1.2.4 generics_0.1.3
## [4] lattice_0.22-6 RSQLite_2.3.7 digest_0.6.37
## [7] magrittr_2.0.3 grid_4.4.1 evaluate_1.0.1
## [10] bookdown_0.40 fastmap_1.2.0 blob_1.2.4
## [13] Matrix_1.7-0 jsonlite_1.8.9 DBI_1.2.3
## [16] BiocManager_1.30.25 httr_1.4.7 fansi_1.0.6
## [19] textshaping_0.4.0 jquerylib_0.1.4 cli_3.6.3
## [22] rlang_1.1.4 dbplyr_2.5.0 basilisk.utils_1.16.0
## [25] bit64_4.5.2 withr_3.0.1 cachem_1.1.0
## [28] yaml_2.3.10 parallel_4.4.1 tools_4.4.1
## [31] dir.expiry_1.12.0 memoise_2.0.1 dplyr_1.1.4
## [34] filelock_1.0.3 basilisk_1.16.0 BiocGenerics_0.50.0
## [37] reticulate_1.39.0 curl_5.2.3 png_0.1-8
## [40] vctrs_0.6.5 R6_2.5.1 BiocFileCache_2.12.0
## [43] lifecycle_1.0.4 fs_1.6.4 htmlwidgets_1.6.4
## [46] bit_4.5.0 ragg_1.3.3 pkgconfig_2.0.3
## [49] desc_1.4.3 pkgdown_2.1.1 pillar_1.9.0
## [52] bslib_0.8.0 Rcpp_1.0.13 glue_1.8.0
## [55] systemfonts_1.1.0 highr_0.11 xfun_0.48
## [58] tibble_3.2.1 tidyselect_1.2.1 knitr_1.48
## [61] htmltools_0.5.8.1 rmarkdown_2.28 compiler_4.4.1