B1 Acquiring and exploring 3202 1KG genotypes from chr17 with T2T reference
Vincent J. Carey, stvjc at channing.harvard.edu
April 04, 2023
B1_1KG_chr17.RmdObjectives
This package demonstrates the use of genotypes obtained via calls against the T2T reference genome. Our objectives are:
- acquire and make the genotypes accessible to R/Bioconductor
- bind relevant sample-level metadata to the resource
- acquire associated RNA-seq assay outputs for a subset of the genotyped samples
- perform a series of GWAS to identify expression-based quantitative trait loci (eQTL)
Acquiring 3202 human genotypes called against the T2T reference
The cloud resource; environment variable setting
We have made a large resource available in the hail.is MatrixTable format. This can be retrieved without egress charges using the code in this vignette. A convenient approach uses rclone, as described in the appendix. The zipped MatrixTable folders are at
https://mghp.osn.xsede.org/bir190004-bucket01/Bioc1KGt2t/t17.zipThis 42GB file should be unzipped in a folder to which the environment variable HAIL_T2T_CHR17 will point.
Initial checks of the resource
Ensure that Sys.getenv("HAIL_T2T_CHR17") returns the location of the unzipped MatrixTable resource.
library(BiocT2T)
library(BiocHail)
hl = hail_init()
# NB the following two commands are now encapsulated in the rg_update function
nn <- hl$get_reference('GRCh38')
nn <- nn$read(system.file("json/t2tAnVIL.json", package="BiocHail"))
# updates the hail reference genome
bigloc = Sys.getenv("HAIL_T2T_CHR17")
if (nchar(bigloc)>0) {
mt17 <- hl$read_matrix_table(Sys.getenv("HAIL_T2T_CHR17"))
mt17$count()
}## [[1]]
## [1] 3824434
##
## [[2]]
## [1] 3202Population stratification assessment via PCA
PCs in R matrices
The following computation takes close to 30 minutes with 64 cores. We saved the loadings in pc_990kloci.
pcastuff = hl$hwe_normalized_pca(mt17$sample_rows(.25)$GT)
data(pc_990kloci)
pc_990kloci[1:3,1:4]## PC1 PC2 PC3 PC4
## HG00096 0.08571635 -0.09670515 -0.04432893 0.032206907
## HG00097 0.07130168 -0.06820003 -0.01072185 0.004602563
## HG00099 0.06578964 -0.08644191 -0.01452058 -0.010706626PCA has also been performed with samples of 3000 and 190000 loci.
Annotation of samples
The populations of origin of the 3202 samples are tabulated in igsr_pops.
##
## AFR AMR EAS EUR SAS
## 183 10 4 5 5 5
igsr_pops |> dplyr::filter(`Superpopulation code` %in% c("AFR", "SAS")) |>
dplyr::select(`Superpopulation name`, `Population code`, `Population name`) |>
dplyr::arrange(desc(`Superpopulation name`))## Superpopulation name Population code Population name
## 1 South Asian Ancestry BEB Bengali
## 2 South Asian Ancestry PJL Punjabi
## 3 South Asian Ancestry GIH Gujarati
## 4 South Asian Ancestry STU Tamil
## 5 South Asian Ancestry ITU Telugu
## 6 African Ancestry ACB African Caribbean
## 7 African Ancestry GWW Gambian Wolof
## 8 African Ancestry ASW African Ancestry SW
## 9 African Ancestry YRI Yoruba
## 10 African Ancestry GWD Gambian Mandinka
## 11 African Ancestry MSL Mende
## 12 African Ancestry ESN Esan
## 13 African Ancestry LWK Luhya
## 14 African Ancestry GWF Gambian Fula
## 15 African Ancestry GWJ Gambian JolaLatitude and longitude are also available for the population centers from which samples were collected.
Visualizing genetic variation
We have several approaches to static visualization of PCs.
A pairs plot for selected PCs for superpopulation visualization based on 190000 loci can be quickly produced.

A more carefully annotated view is produced using GGally::ggpairs:
strat2d(pc_190kloci, 1:4, superOnly=TRUE, GGally=TRUE)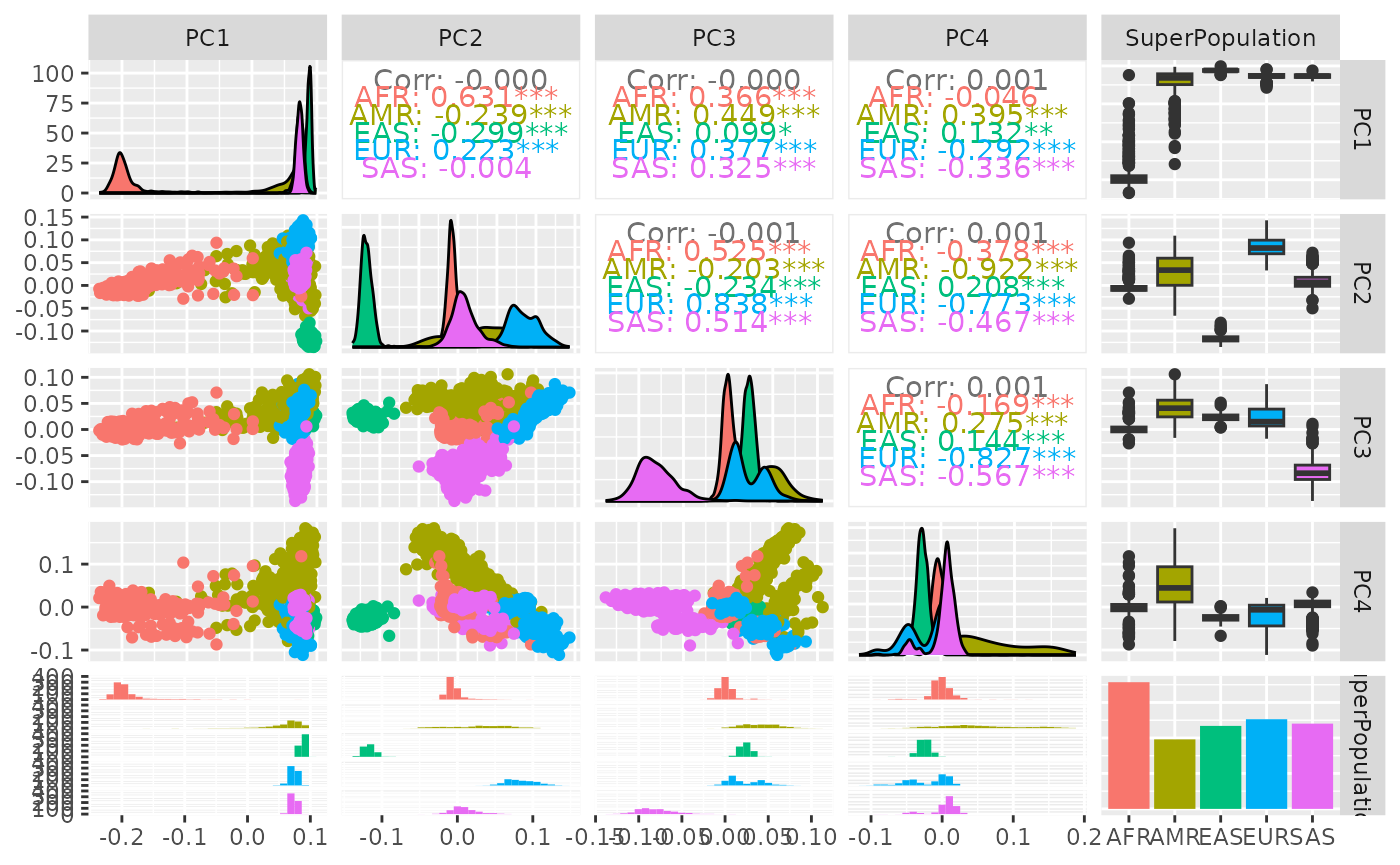
Appendix: Using rclone with docker to get the chr17 data
It can be painful to install and configure rclone. We use a docker container. Let RC_DATADIR be an environment variable evaluating to an available folder.
Also, place the text file with contents
[osn]
type = s3
provider = AWS
endpoint = https://mghp.osn.xsede.org
acl = public
no_check_bucket = truein a file rclone.conf in a folder pointed to by the environment variable RC_CONFDIR.
Then the following
docker run -v $RC_DATADIR:/data -v $RC_CONFDIR:/config/rclone -t rclone/rclone:latest ls osn:/bir190004-bucket01/Bioc1KGt2twill list the files with 1KG samples genotyped against the T2T reference.
Use the rclone copyto command to obtain a local copy of the zip file t17.zip in the folder pointed to by $RC_DATADIR:
docker run -v $RC_DATADIR:/data -v $RC_CONFDIR:/config/rclone -t rclone/rclone:latest copyto osn:/bir190004-bucket01/Bioc1KGt2t/t17.zip ./t17.zipThis file should be unzipped in a folder to which the environment variable HAIL_T2T_CHR17 will point.