scvi-tools CITE-seq tutorial in R, using serialized tutorial components
Follows scvi-tools doc, Gayoso, Steier et al. DOI 10.1038/s41592-020-01050-x
September 26, 2023
Source:vignettes/citeseq_tut.Rmd
citeseq_tut.RmdA CITE-seq example
The purpose of this vignette is to illustrate the feasibility of reflecting the material in the online tutorial for scvi-tools 0.20.0 in Bioconductor. The authors of the tutorial describe it as producing
a joint latent representation of cells, denoised data for both protein and RNA
Additional tasks include
integrat[ing] datasets, and comput[ing] differential expression of RNA and protein
The integration concerns the simultaneous analysis of two datasets from 10x genomcs.
In this vignette we carry out the bulk of the tutorial activities using R and Bioconductor, reaching to scvi-tools python code via basilisk.
Retrieval of PBMC data
The following chunk will acquire (and cache, using BiocFileCache) a preprocessed version of the 10k and 5k combined CITE-seq experiments from the scvi-tools data repository.
## AnnData object with n_obs × n_vars = 10849 × 4000
## obs: 'n_genes', 'percent_mito', 'n_counts', 'batch', '_scvi_labels', '_scvi_batch'
## var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
## uns: '_scvi_manager_uuid', '_scvi_uuid', 'hvg', 'log1p'
## obsm: 'protein_expression'
## layers: 'counts'Retrieval of fitted VAE
The totalVI variational autoencoder was fit with these data. A fitted version is retrieved and cached using
vae = getCiteseqTutvae()## INFO File /home/vincent/TEMP/Rtmp8FzFo7/vae2_ov/model.pt already downloaded
## INFO Computing empirical prior initialization for protein background.This is an instance of an S3 class, python.builtin.object, defined in the reticulate package.
class(vae)## [1] "scvi.model._totalvi.TOTALVI"
## [2] "scvi.model.base._rnamixin.RNASeqMixin"
## [3] "scvi.model.base._vaemixin.VAEMixin"
## [4] "scvi.model.base._archesmixin.ArchesMixin"
## [5] "scvi.model.base._base_model.BaseModelClass"
## [6] "scvi.autotune._types.TunableMixin"
## [7] "python.builtin.object"Some fields of interest that are directly available from the instance include an indicator of the trained state, the general parameters used to train, and the “anndata” (annotated data) object that includes the input counts and various results of preprocessing:
vae$is_trained## [1] TRUE
cat(vae$`_model_summary_string`)## TotalVI Model with the following params:
## n_latent: 20, gene_dispersion: gene, protein_dispersion: protein, gene_likelihood: nb, latent_distribution: normal
vae$adata## AnnData object with n_obs × n_vars = 10849 × 4000
## obs: 'n_genes', 'percent_mito', 'n_counts', 'batch', '_scvi_labels', '_scvi_batch'
## var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
## uns: '_scvi_manager_uuid', '_scvi_uuid', 'hvg', 'log1p'
## obsm: 'protein_expression'
## layers: 'counts'The structure of the VAE is reported using
vae$moduleThis is quite voluminous and is provided in an appendix.
Trace of (negative) ELBO values
The negative “evidence lower bound” (ELBO) is a criterion that is minimized in order to produce a fitted autoencoder. The scvi-tools totalVAE elgorithm creates a nonlinear projection of the inputs to a 20-dimensional latent space, and a decoder that transforms object positions in the latent space to positions in the space of observations that are close to the original input positions.
The negative ELBO values are computed for samples from the training data and for “left out” validation samples. Details on the validation assessment would seem to be part of pytorch lightning. More investigation of scvi-tools code and doc are in order.
h = vae$history
npts = nrow(h$elbo_train)
plot(seq_len(npts), as.numeric(h$elbo_train[[1]]), ylim=c(1200,1400),
type="l", col="blue", main="Negative ELBO over training epochs",
ylab="neg. ELBO", xlab="epoch")
graphics::legend(300, 1360, lty=1, col=c("blue", "orange"), legend=c("training", "validation"))
graphics::lines(seq_len(npts), as.numeric(h$elbo_validation[[1]]), type="l", col="orange")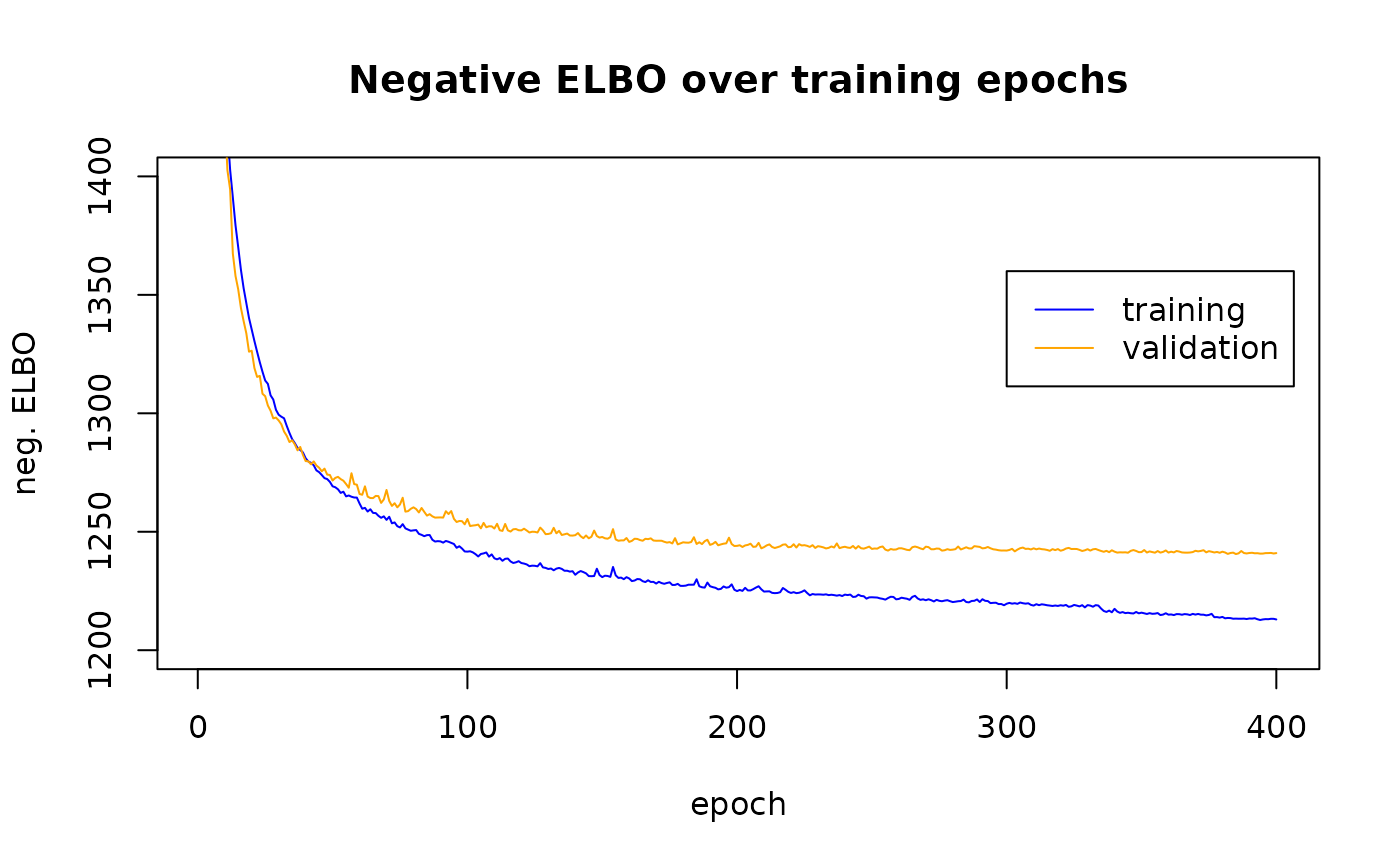
Normalized quantities
On a CPU, the following can take a long time.
NE = vae$get_normalized_expression(n_samples=25L,
return_mean=TRUE,
transform_batch=c("PBMC10k", "PBMC5k")
)We provide the totalVI-based denoised quantities in
denoised = getTotalVINormalized5k10k()
vapply(denoised, dim, integer(2))## rna_nmlzd prot_nmlzd
## [1,] 10849 10849
## [2,] 4000 14Note that these have features as columns, samples (cells) as rows.
## [1] "AL645608.8" "HES4" "ISG15" "TTLL10" "TNFRSF18"
## [6] "TNFRSF4"## [1] "CD3_TotalSeqB" "CD4_TotalSeqB" "CD8a_TotalSeqB" "CD14_TotalSeqB"
## [5] "CD15_TotalSeqB" "CD16_TotalSeqB"UMAP projection of Leiden clustering in the totalVI latent space
We have stored a fully loaded anndata instance for retrieval to inspect the latent space and clustering produced by the tutorial notebook procedure. The images produced here do not agree exactly with what I see in the colab pages for 0.20.0. The process was run in Jetstream2, not in colab.
full = getTotalVI5k10kAdata()
# class distribution
cllabs = full$obs$leiden_totalVI
blabs = full$obs$batch
table(cllabs)## cllabs
## 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
## 2287 1899 1138 866 787 660 637 587 461 375 334 261 260 208 59 19
## 16
## 11
um = full$obsm$get("X_umap")
dd = data.frame(umap1=um[,1], umap2=um[,2], clust=factor(cllabs), batch=blabs)
ggplot(dd, aes(x=umap1, y=umap2, colour=clust)) + geom_point(size=.05) +
guides(color = guide_legend(override.aes = list(size = 4)))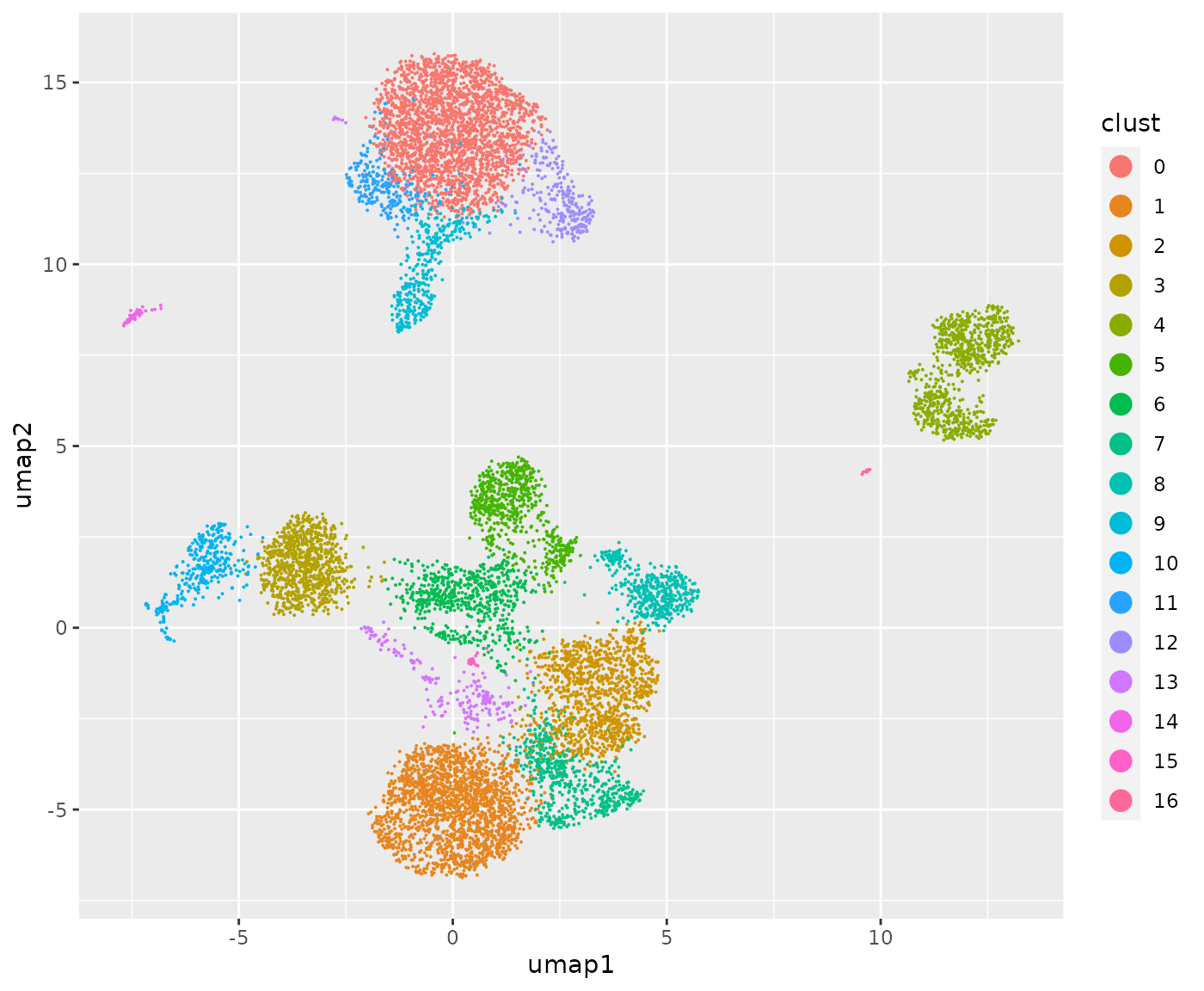
Effectiveness at accommodating the two-batch design is suggested by the mixed representation of the batches in all the Leiden clusters.
ggplot(dd, aes(x=umap1, y=umap2, colour=factor(batch))) + geom_point(size=.05)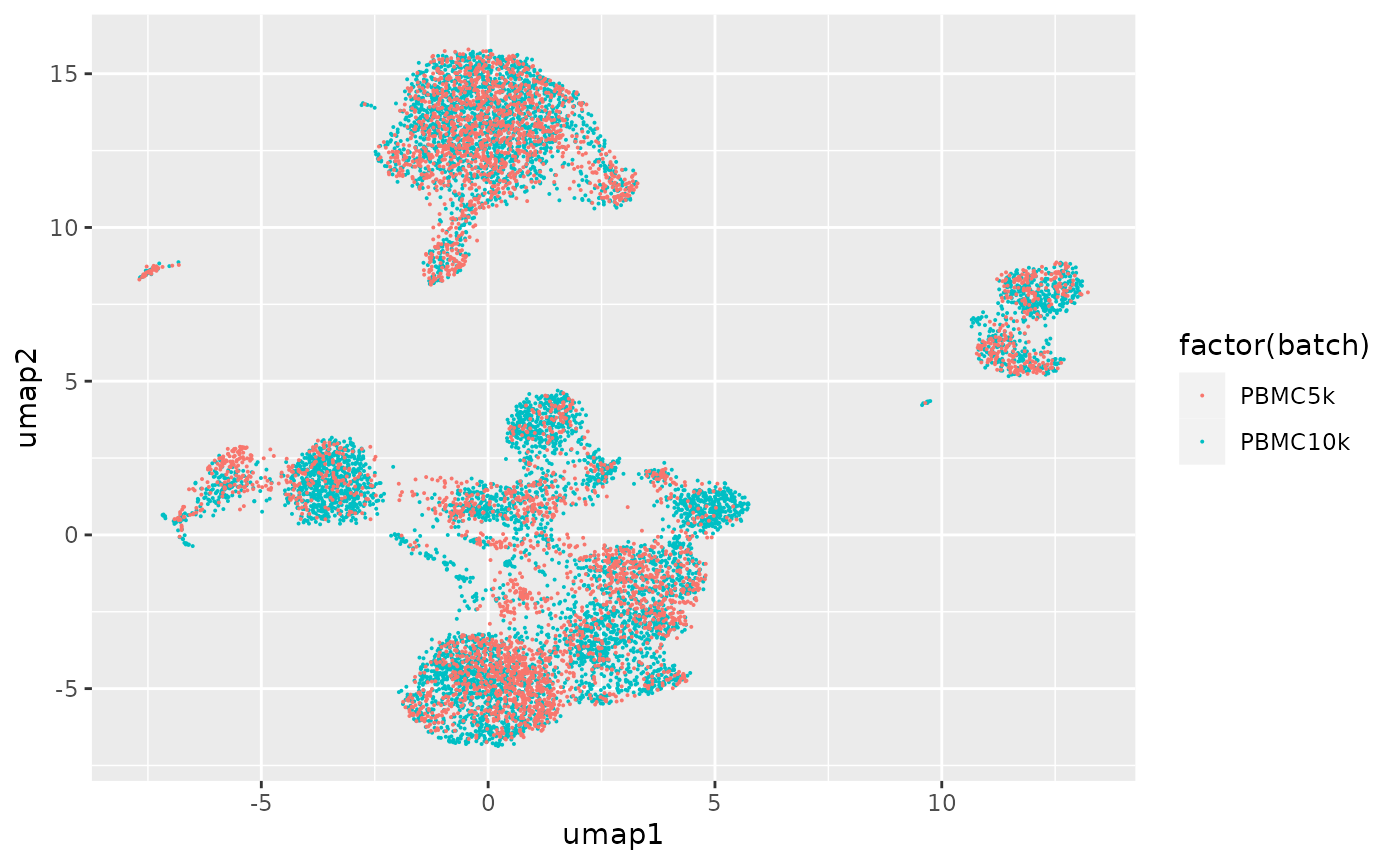
Protein abundances in projected clusters
We focus on four of the ADT. Points (cells) in the UMAP projection given above are colored by the estimated abundance of the proteins quantified via (normalized) ADT abundance. Complementary expression of CD4 and CD8a is suggested by the configurations in the middle two panels.
pro4 = denoised$prot_nmlzd[,1:4]
names(pro4) = gsub("_.*", "", names(pro4))
wprot = cbind(dd, pro4)
mm = melt(wprot, id.vars=c("clust", "batch", "umap1", "umap2"))
utils::head(mm,3)## clust batch umap1 umap2 variable value
## 1 0 PBMC10k 1.0286884 14.58188 CD3 0.6282051
## 2 0 PBMC10k 0.8443313 15.29502 CD3 11.9159498
## 3 0 PBMC10k 0.8057814 13.96269 CD3 8.9384489
ggplot(mm, aes(x=umap1, y=umap2, colour=log1p(value))) +
geom_point(size=.1) + facet_grid(.~variable)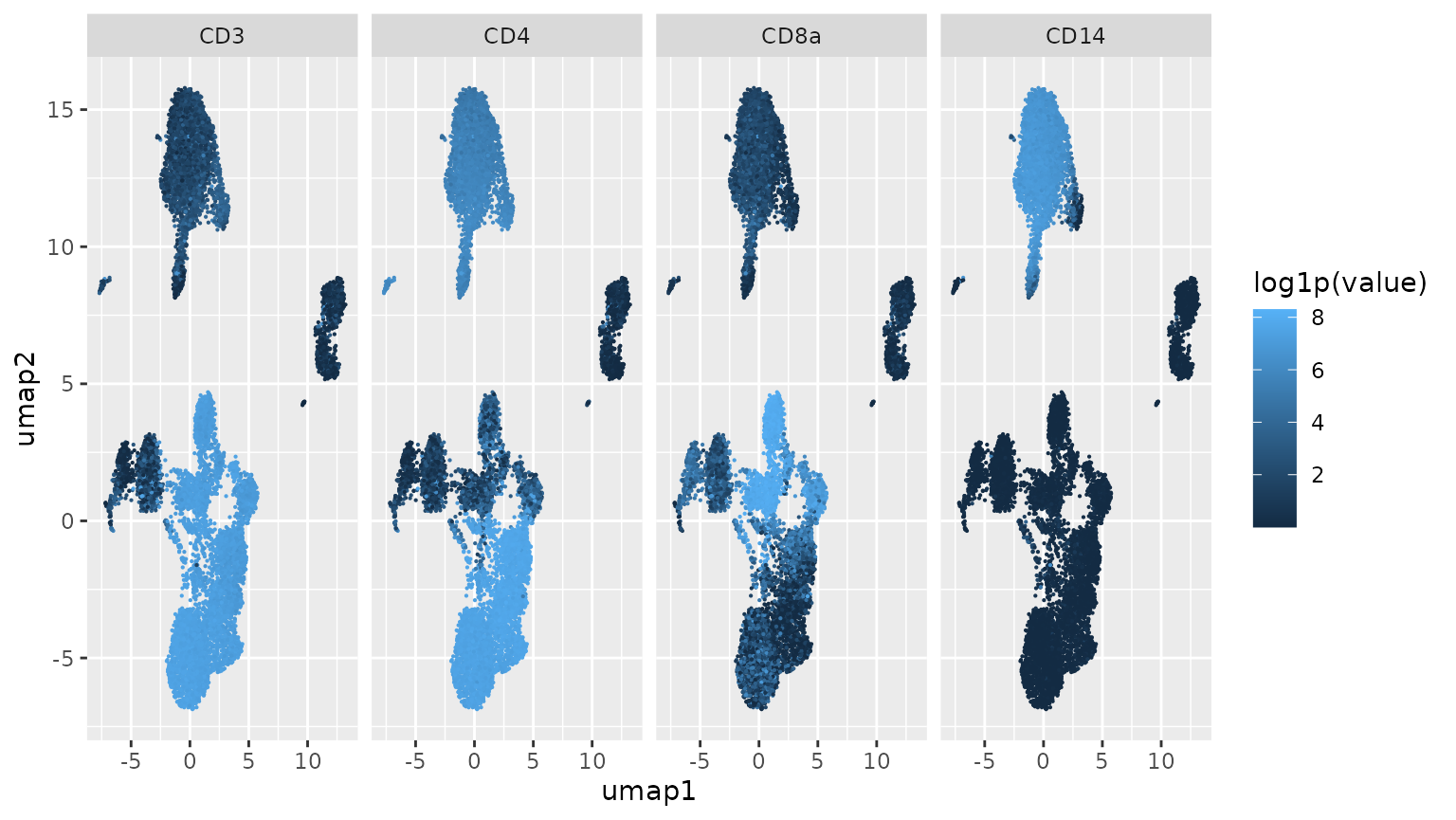
Conclusions
We have shown that all the results of the totalVI application in the tutorial are readily accessible with utilities in scviR. Additional work on details of differential expression are present in the tutorial and can be explored by the interested reader/user.
Appendix: The VAE module
The structure of the VAE is reported using
vae$module## TOTALVAE(
## (encoder): EncoderTOTALVI(
## (encoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=4016, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## (Layer 1): Sequential(
## (0): Linear(in_features=258, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## )
## )
## (z_mean_encoder): Linear(in_features=256, out_features=20, bias=True)
## (z_var_encoder): Linear(in_features=256, out_features=20, bias=True)
## (l_gene_encoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=4016, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## )
## )
## (l_gene_mean_encoder): Linear(in_features=256, out_features=1, bias=True)
## (l_gene_var_encoder): Linear(in_features=256, out_features=1, bias=True)
## )
## (decoder): DecoderTOTALVI(
## (px_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=22, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## )
## )
## (px_scale_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=278, out_features=4000, bias=True)
## (1): None
## (2): None
## (3): None
## (4): None
## )
## )
## )
## (px_scale_activation): Softmax(dim=-1)
## (py_back_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=22, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## )
## )
## (py_back_mean_log_alpha): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=278, out_features=14, bias=True)
## (1): None
## (2): None
## (3): None
## (4): None
## )
## )
## )
## (py_back_mean_log_beta): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=278, out_features=14, bias=True)
## (1): None
## (2): None
## (3): None
## (4): None
## )
## )
## )
## (py_fore_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=22, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## )
## )
## (py_fore_scale_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=278, out_features=14, bias=True)
## (1): None
## (2): None
## (3): ReLU()
## (4): None
## )
## )
## )
## (sigmoid_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=22, out_features=256, bias=True)
## (1): BatchNorm1d(256, eps=0.001, momentum=0.01, affine=True, track_running_stats=True)
## (2): None
## (3): ReLU()
## (4): Dropout(p=0.2, inplace=False)
## )
## )
## )
## (px_dropout_decoder_gene): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=278, out_features=4000, bias=True)
## (1): None
## (2): None
## (3): None
## (4): None
## )
## )
## )
## (py_background_decoder): FCLayers(
## (fc_layers): Sequential(
## (Layer 0): Sequential(
## (0): Linear(in_features=278, out_features=14, bias=True)
## (1): None
## (2): None
## (3): None
## (4): None
## )
## )
## )
## )
## )Session information
utils::sessionInfo()## R version 4.3.1 Patched (2023-08-27 r85021)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.3 LTS
##
## Matrix products: default
## BLAS: /home/vincent/R-4-3-dist/lib/R/lib/libRblas.so
## LAPACK: /home/vincent/R-4-3-dist/lib/R/lib/libRlapack.so; LAPACK version 3.11.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: America/New_York
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics utils stats4 methods base
##
## other attached packages:
## [1] reshape2_1.4.4 ggplot2_3.4.3
## [3] scviR_1.1.1 SingleCellExperiment_1.23.0
## [5] SummarizedExperiment_1.31.1 Biobase_2.61.0
## [7] GenomicRanges_1.53.1 GenomeInfoDb_1.37.4
## [9] IRanges_2.35.2 S4Vectors_0.39.1
## [11] BiocGenerics_0.47.0 MatrixGenerics_1.13.1
## [13] matrixStats_1.0.0 shiny_1.7.5
## [15] basilisk_1.13.1 BiocStyle_2.29.1
##
## loaded via a namespace (and not attached):
## [1] DBI_1.1.3 bitops_1.0-7
## [3] gridExtra_2.3 rlang_1.1.1
## [5] magrittr_2.0.3 scater_1.29.4
## [7] compiler_4.3.1 RSQLite_2.3.1
## [9] dir.expiry_1.9.0 DelayedMatrixStats_1.23.4
## [11] png_0.1-8 systemfonts_1.0.4
## [13] vctrs_0.6.3 stringr_1.5.0
## [15] pkgconfig_2.0.3 crayon_1.5.2
## [17] fastmap_1.1.1 dbplyr_2.3.3
## [19] XVector_0.41.1 ellipsis_0.3.2
## [21] labeling_0.4.3 scuttle_1.11.2
## [23] utf8_1.2.3 promises_1.2.1
## [25] rmarkdown_2.24 grDevices_4.3.1
## [27] ggbeeswarm_0.7.2 ragg_1.2.5
## [29] purrr_1.0.2 bit_4.0.5
## [31] xfun_0.40 zlibbioc_1.47.0
## [33] cachem_1.0.8 beachmat_2.17.16
## [35] jsonlite_1.8.7 blob_1.2.4
## [37] later_1.3.1 DelayedArray_0.27.10
## [39] BiocParallel_1.35.4 irlba_2.3.5.1
## [41] parallel_4.3.1 R6_2.5.1
## [43] RColorBrewer_1.1-3 bslib_0.5.1
## [45] stringi_1.7.12 limma_3.57.7
## [47] reticulate_1.32.0 jquerylib_0.1.4
## [49] Rcpp_1.0.11 bookdown_0.35
## [51] knitr_1.44 httpuv_1.6.11
## [53] Matrix_1.6-1 tidyselect_1.2.0
## [55] viridis_0.6.4 abind_1.4-5
## [57] yaml_2.3.7 codetools_0.2-19
## [59] curl_5.0.2 plyr_1.8.8
## [61] lattice_0.21-8 tibble_3.2.1
## [63] withr_2.5.0 basilisk.utils_1.13.3
## [65] evaluate_0.21 desc_1.4.2
## [67] BiocFileCache_2.9.1 pillar_1.9.0
## [69] BiocManager_1.30.22 filelock_1.0.2
## [71] generics_0.1.3 rprojroot_2.0.3
## [73] RCurl_1.98-1.12 munsell_0.5.0
## [75] sparseMatrixStats_1.13.4 scales_1.2.1
## [77] xtable_1.8-4 glue_1.6.2
## [79] pheatmap_1.0.12 tools_4.3.1
## [81] datasets_4.3.1 BiocNeighbors_1.19.0
## [83] ScaledMatrix_1.9.1 fs_1.6.3
## [85] grid_4.3.1 colorspace_2.1-0
## [87] GenomeInfoDbData_1.2.10 beeswarm_0.4.0
## [89] BiocSingular_1.17.1 vipor_0.4.5
## [91] cli_3.6.1 rsvd_1.0.5
## [93] textshaping_0.3.6 fansi_1.0.4
## [95] viridisLite_0.4.2 S4Arrays_1.1.6
## [97] dplyr_1.1.3 gtable_0.3.4
## [99] sass_0.4.7 digest_0.6.33
## [101] ggrepel_0.9.3 SparseArray_1.1.12
## [103] farver_2.1.1 memoise_2.0.1
## [105] htmltools_0.5.6 pkgdown_2.0.7
## [107] lifecycle_1.0.3 httr_1.4.7
## [109] statmod_1.5.0 mime_0.12
## [111] bit64_4.0.5